Search results
Search for "force fields" in Full Text gives 25 result(s) in Beilstein Journal of Nanotechnology.
Multiscale modelling of biomolecular corona formation on metallic surfaces
Beilstein J. Nanotechnol. 2024, 15, 215–229, doi:10.3762/bjnano.15.21

- GROMACS-2018.6 and PLUMED (PLUMED2-2.5.1.conda.5) software packages [29][30][31]. CHARMM-GUI/Nanomaterial Modeler was employed to construct the topology and force fields of three fcc surfaces of Al: (100), (110), and (111) [32]. The General Amber Force Field (GAFF) was utilized to model side-chains









Ultralow-energy amorphization of contaminated silicon samples investigated by molecular dynamics
Beilstein J. Nanotechnol. 2023, 14, 834–849, doi:10.3762/bjnano.14.68

- amorphization. We will also show that favorable angles to minimize the implantation of the species of the contamination layer are in the range of 60° to 75°. Computational Methods Force fields The force fields used to simulate the Ar bombardment of a contaminated silicon sample have already been described in a
- Sycheva et al. agree well with our values, while the other values are well above. The difference in sputtering yields between our results and the literature data can be explained by the difference between the ReaxFF potential used in our simulations and the force fields or experimental methods used in the










Transferability of interatomic potentials for silicene
Beilstein J. Nanotechnol. 2023, 14, 574–585, doi:10.3762/bjnano.14.48

- –Weber, EDIP, ReaxFF, COMB, and machine-learning-based interatomic potentials. A quantitative systematic comparison and a discussion of the results obtained are reported. Keywords: 2D materials; DFT; force fields; interatomic potentials; mechanical properties; silicene; Introduction We are living in

![[Graphic 3]](/bjnano/content/inline/2190-4286-14-48-i7.svg?max-width=637&scale=1.18182)

Influence of water contamination on the sputtering of silicon with low-energy argon ions investigated by molecular dynamics simulations
Beilstein J. Nanotechnol. 2022, 13, 986–1003, doi:10.3762/bjnano.13.86

- [30]. ReaxFF force fields are specifically tuned for a set of atomic interactions. They are developed from quantum calculations and are adapted for MD simulations, providing faster calculations than pure quantum electrodynamics (QED)/density functional theory (DFT) and more information than classical
- Methods Force fields The ReaxFF force field differs from other established force fields as it aims to bridge quantum mechanics (QM) and classical MD. Quantum mechanics algorithms are limited to small-sized samples (up to a few hundreds of atoms) due to the difficulty to solve Schrödinger equations for



















Effect of lubricants on the rotational transmission between solid-state gears
Beilstein J. Nanotechnol. 2022, 13, 54–62, doi:10.3762/bjnano.13.3

- MD simulations. For the force fields, we choose the adaptive intermolecular reactive empirical bond order (AIREBO) potential [59]. This potential was designed for hydrocarbon systems and can reach reasonable densities for the molecules we will use later. We have used two different protocols. For




Irradiation-driven molecular dynamics simulation of the FEBID process for Pt(PF3)4
Beilstein J. Nanotechnol. 2021, 12, 1151–1172, doi:10.3762/bjnano.12.86

- deposition; irradiation-driven molecular dynamics; irradiation-induced chemistry; platinum nanostructures; reactive force fields; Introduction The controllable fabrication of nanostructures with nanoscale resolution remains a considerable scientific and technological challenge [1]. To address this challenge
- of atomic partial charges, or alteration of interatomic interactions) are simulated by means of MD with reactive force fields [17][18] using the advanced software packages MBN Explorer [19] and MBN Studio [20]. MBN Explorer is a multi-purpose software package for multiscale simulations of structure












Molecular dynamics modeling of the influence forming process parameters on the structure and morphology of a superconducting spin valve
Beilstein J. Nanotechnol. 2020, 11, 1776–1788, doi:10.3762/bjnano.11.160

- the nanosystem. There are many possible choices for the type of potential, but recently, due to its accuracy and adequacy, many-particle force fields have gained great popularity. In this work, we used the potential of the modified embedded-atom method (MEAM) which is based on density functional











Calculating free energies of organic molecules on insulating substrates
Beilstein J. Nanotechnol. 2017, 8, 667–674, doi:10.3762/bjnano.8.71

- significantly lower the adhesion energy of molecules at step edges and cannot be ignored when considering film growth at step edges and terraces. Methods Classical force fields In order to calculate free energies of molecular processes on surfaces, long-timescale MD simulations are needed. Therefore classical
- force fields are used since the computational cost associated with ab initio methods is too high. The LAMMPS code was used for all calculations [44] along with a combination of several classical force fields. A Buckingham potential was used to describe the interactions inside the KCl slab, as






Noise in NC-AFM measurements with significant tip–sample interaction
Beilstein J. Nanotechnol. 2016, 7, 1885–1904, doi:10.3762/bjnano.7.181
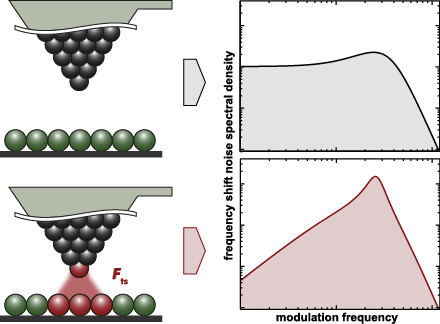
- science tool, especially when it comes to studying non-conducting surfaces [3][4], to map sub-molecular structures [5] or to measure forces [6] and force fields [7] with highest resolution. The primary imaging signal in NC-AFM is the frequency shift Δf of a probe resonator carrying a tip interacting with







![[Graphic 32]](/bjnano/content/inline/2190-4286-7-181-i73.png?max-width=637&scale=1.18182) wit...
wit...

![[Graphic 34]](/bjnano/content/inline/2190-4286-7-181-i75.png?max-width=637&scale=1.18182) wit...
wit...





Numerical investigation of depth profiling capabilities of helium and neon ions in ion microscopy
Beilstein J. Nanotechnol. 2016, 7, 1749–1760, doi:10.3762/bjnano.7.168

- dynamics (MD) simulations, possible changes in the sample upon irradiation due to chemical reactions, as they are included in reactive force fields [27][28][29], are not contained in the model, but SD_TRIM_SP has the advantage to allow for the simulation of high fluences up in the 1018 ions/cm2 range, i.e










Multiscale modeling of lithium ion batteries: thermal aspects
Beilstein J. Nanotechnol. 2015, 6, 987–1007, doi:10.3762/bjnano.6.102

- utilizing classical force fields to continuum theories. But even within continuum theories it is for practical reasons important to distinguish between microstructure-resolved and porous-electrode theories to develop consistent theories for both scales. The knowledge of the material parameters and their
- ) simulations once the force fields for the interaction between the molecules are known [14][15]. Information on interface properties can be obtained from MD simulations and from DFT simulations [15]. MD simulations are especially relevant to study the solvation properties of the ions [15][16][17], which are








Nanoparticle shapes by using Wulff constructions and first-principles calculations
Beilstein J. Nanotechnol. 2015, 6, 361–368, doi:10.3762/bjnano.6.35

- of atoms are presently limited to atoms with few valence electrons, such as simple metals or semiconductors [50][51][52]. Late transition metals are typically simulated by using advanced force-fields or other approaches [53][54][55]. The advantage of the atomistic Wulff construction is that one




Multifunctional layered magnetic composites
Beilstein J. Nanotechnol. 2015, 6, 134–148, doi:10.3762/bjnano.6.13

- in aqueous solution by using empirical force fields [44][45][63][64]. Investigation of biologically designed metal-specific chelators for potential metal recovery and waste remediation applications [65], and the Kawska–Zahn docking procedure were described previously [43]. Along this line, ion













Double layer effects in a model of proton discharge on charged electrodes
Beilstein J. Nanotechnol. 2014, 5, 973–982, doi:10.3762/bjnano.5.111

- reactive force field procedure to statistically study the large number of proton transfer pathways by developing empirical valence bond (EVB) force fields for Grotthuss style proton migration and proton discharge at the water/Pt(111) [16][17] and the water/Ag(111) interface [18]. The first EVB models were






Molecular dynamics simulations of mechanical failure in polymorphic arrangements of amyloid fibrils containing structural defects
Beilstein J. Nanotechnol. 2013, 4, 429–440, doi:10.3762/bjnano.4.50

- modifications. Molecular dynamics simulations Molecular dynamics simulations were run using AMBER9 [35] and NAMD2.7b1 [36] simulation packages, with the all atom AMBER99SB [37] and the CHARMM22/CMAP [38] force fields used, respectively. All models were explicitly solvated in a periodic water-box of TIP3









Influence of the solvent on the stability of bis(terpyridine) structures on graphite
Beilstein J. Nanotechnol. 2013, 4, 269–277, doi:10.3762/bjnano.4.29

- model system, the solvation of a bis(terpyridine) isomer in water and 1,2,4-trichlorobenzene was studied with an explicit solvation model. The inclusion of solvation has a noticeable effect on adsorption energies. Although the results of the various considered force fields differ quite significantly
- of the considered systems and the requirement to perform thermal averages make first-principles electronic-structure calculations computationally prohibitively expensive. Therefore we employed classical force fields as included in the Forcite module of the Accelrys’ Materials Studio package to
- describe the interaction between adsorbate, substrate and solvent. It is true that the force fields in this package tend to overestimate BTP adsorption energies on graphite [12]. Still, trends in the stability of BTP stuctures on graphite as a function of the environment should still be reproduced. As a









Bimodal atomic force microscopy driving the higher eigenmode in frequency-modulation mode: Implementation, advantages, disadvantages and comparison to the open-loop case
Beilstein J. Nanotechnol. 2013, 4, 198–207, doi:10.3762/bjnano.4.20

- out integration of the force gradient to calculate the forces as a function of the xyz-coordinates. This results in three dimensional force fields, as they are usually obtained in time-consuming volume-scanning applications [33][34]. Although the approach described in [31][32] is not yet






Advanced atomic force microscopy techniques
Beilstein J. Nanotechnol. 2012, 3, 893–894, doi:10.3762/bjnano.3.99

- . Data acquisition times have already reached the millisecond range, enabling the visualization of the dynamic behavior of biological molecules and cells. Other recent accomplishments include imaging of organic molecules with unprecedented resolution, full three-dimensional mapping of surface force
- fields, and the imaging and discrimination of individual chemical bonds. The development of advanced techniques is the focus of this Thematic Series, following the Thematic Series “Scanning probe microscopy and related techniques” edited by Ernst Meyer and the Thematic Series “Noncontact atomic force
Probing three-dimensional surface force fields with atomic resolution: Measurement strategies, limitations, and artifact reduction
Beilstein J. Nanotechnol. 2012, 3, 637–650, doi:10.3762/bjnano.3.73

- interpretation. In this paper, we analyze their impact on the acquired data, compare different methods to record atomic-resolution surface force fields, and determine the approaches that suffer the least from the associated artifacts. The related discussion underscores the idea that since force fields recorded
- sample surface. Force fields have now been recorded on NiO(001) [10][12][13], MgO/Ag(001) [14], NaCl(001) [15][16], Si(111)-(7×7) [17][18][19], HOPG [20][21], KBr(001) [9][22][23], Cu(111) [24], and CaCO3() [25] surfaces, as well as single molecules of PTCDA [26][27], pentacene [28], CO [29], C60 [30
- –mica interface [35]. The methods most frequently reported in the literature to record two- and three-dimensional force fields above sample surfaces may be divided into two general categories (Figure 1): 1) The curve-by-curve method, in which individual curves of frequency shift versus tip–sample








Models of the interaction of metal tips with insulating surfaces
Beilstein J. Nanotechnol. 2012, 3, 329–335, doi:10.3762/bjnano.3.37

- tip–surface combinations, the calculated force fields would result in the anions being imaged as prominent protrusions in a constant-frequency-shift image of the surfaces. To demonstrate this, and show the extent of typical atomic scale corrugation, we simulated the imaging of the NaCl surface with






Graphite, graphene on SiC, and graphene nanoribbons: Calculated images with a numerical FM-AFM
Beilstein J. Nanotechnol. 2012, 3, 301–311, doi:10.3762/bjnano.3.34

- future. It should be mentioned also that when the tip interacts chemically with the substrate through bond creation between the tip apex atom and surface atoms, the choice of the force-field method may be difficult to justify. In that case, although reactive force fields exist [89][90][91] and may be




Noncontact atomic force microscopy
Beilstein J. Nanotechnol. 2012, 3, 172–173, doi:10.3762/bjnano.3.17
- conference from this series was held in Lindau, Germany, from September 18–22, 2011. Once again, substantial progress was presented; NC-AFM is now able to quantitatively map three-dimensional force fields of surfaces with atomic resolution in ultrahigh vacuum as well as in liquids, and methodological
Self-organizing bioinspired oligothiophene–oligopeptide hybrids
Beilstein J. Nanotechnol. 2011, 2, 525–544, doi:10.3762/bjnano.2.57

- –peptide conjugates and to gain more insight into the structure and dynamical behavior of the aggregates at finite temperatures, a theoretical methodology based on classical mechanical force fields and molecular dynamics simulations was developed [23]. Although molecular models based on classical mechanics

















Towards a scalable and accurate quantum approach for describing vibrations of molecule–metal interfaces
Beilstein J. Nanotechnol. 2011, 2, 427–447, doi:10.3762/bjnano.2.48

- the prediction of vibrational frequencies. From a theoretical point of view, the accuracy of the data obtained from high resolution spectra provides an ideal opportunity to validate the interaction models used to describe the observed molecule, be they empirical force fields or quantum chemical
- conformationally flexible molecules. Due to its conceptual simplicity and the ready availability of reliable empirical force fields (or forces computed ab initio), molecular dynamics is currently the most popular method for determining anharmonic frequencies of large systems (see [7] for an overview of some











Intermolecular vs molecule–substrate interactions: A combined STM and theoretical study of supramolecular phases on graphene/Ru(0001)
Beilstein J. Nanotechnol. 2011, 2, 365–373, doi:10.3762/bjnano.2.42

- of a “hill” site from that of a “valley” position. Table 1 and Table 2 show the differences between these sites for different force fields. Clearly, the calculated binding energies strongly depend on the force field used, as found before in force field calculations addressing 3,3'-BTP adsorption on
- calculated adsorption energy for both the hill and the valley position and the resulting corrugation of the adsorption potential ΔE for PTCDA molecules on graphene/Ru(0001) for different force fields. Dependent on the applied force field, the resulting ΔE ranges from −0.435 to −0.690 eV. To rationalize the
- placed on top of the two different adsorption sites (“hill” and “valley”) and four different force fields were used to optimize the adsorption geometry of the adsorbate (Compass [40], CVFF [41], Dreiding [42], and UFF [43] as implemented in the Accelrys Materials Studio program package). Note that the





